Vorapaxar (Zontivity): Drug Monograph
|
|---|
- Reduction of thrombotic cardiovascular (CV) events in patients with a history of myocardial infarction (MI) or with peripheral arterial disease (PAD)
- Reduction of the rate of a combined endpoint of cardiovascular death, MI, stroke, and urgent coronary revascularization
- One tablet (2.08 mg) once daily by mouth
- Use with aspirin and/or clopidogrel according to their indications or standard of care
-
Do not use vorapaxar in patients with a history of stroke, transient ischemic attack (TIA), or intracranial hemorrhage (ICH); or active pathological bleeding. Antiplatelet agents, including vorapaxar, increase the risk of bleeding, including ICH and fatal bleeding
- Bleeding, including life-threatening and fatal bleeding
- Anemia
- Depression
- Rashes, eruptions, and exanthemas
-
There is no known treatment to reverse the antiplatelet effect of vorapaxar and neither dialysis nor platelet transfusion can be expected to be beneficial if bleeding occurs after overdose.
-
Inhibition of platelet aggregation can be expected for weeks after discontinuation of normal dosing.
-
There is no standard test available to assess the risk of bleeding in an overdose situation.
-
Strong CYP3A inhibitors (e.g., ketoconazole, itraconazole, posaconazole, clarithromycin, nefazodone, ritonavir, saquinavir, nelfinavir, indinavir, boceprevir, telaprevir, telithromycin and conivaptan) - avoid use
-
Strong CYP3A inducers (e.g., rifampin, carbamazepine, St. John's Wort, phenytoin) - avoid use
-
Pregnancy: Pregnancy Category B
-
Labor and Delivery: None
-
Nursing Mothers: It is not known whether vorapaxar or its metabolites are excreted in human milk. Because of the potential for serious adverse reactions in nursing infants, discontinue nursing or discontinue vorapaxar.
-
Renal Impairment: No dose adjustment is required.
-
Hepatic Impairment: No dose adjustment required in patients with mild and moderate impairment. Based on the increased inherent risk of bleeding in patients with severe impairment, vorapaxar is not recommended in such patients.
-
Pediatric Patients: The safety and effectiveness have not been established.
-
Geriatric Patients: No overall differences in safety or effectiveness were observed between elderly patients and younger patients. Since vorapaxar increases the risk of bleeding in proportion to a patient's underlying risk, consider patient age before initiating treatment.
-
It is not known whether vorapaxar or its metabolites are excreted in human milk. Because of the potential for serious adverse reactions in nursing infants, discontinue nursing or discontinue vorapaxar.
-
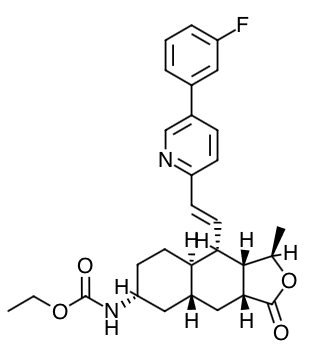
Scientific Name: ethyl [(1R,3aR,4aR,6R,8aR,9S,9aS)-9-{(1E)-2-[5-(3- fluorophenyl)pyridin-2-yl]ethen-1-yl}-1-methyl-3-oxododecahydronaphtho[2,3-c]furan-6-yl]carbamate sulfate
-
Empirical Formula: C29H33FN2O4.H2SO4
-
Molecular Weight: 590.7
-
Vorapaxar is a reversible antagonist of the protease-activated receptor-1 (PAR-1) expressed on platelets, but its long half-life makes it effectively irreversible. Vorapaxar inhibits thrombin-induced and thrombin receptor agonist peptide (TRAP)-induced platelet aggregation in in vitro studies. Vorapaxar does not inhibit platelet aggregation induced by adenosine diphosphate (ADP), collagen or a thromboxane mimetic and does not affect coagulation parameters ex vivo. PAR-1 receptors are also expressed in a wide variety of cell types, including endothelial cells, neurons, and smooth muscle cells, but the pharmacodynamic effects of vorapaxar in these cell types have not been assessed.
-
At the recommended dose, vorapaxar achieves ≥80% inhibition of TRAP-induced platelet aggregation within one week of initiation of treatment. The duration of platelet inhibition is dose- and concentration-dependent. Inhibition of TRAP-induced platelet aggregation at a level of 50% can be expected at 4 weeks after discontinuation of daily doses of vorapaxar 2.08 mg, consistent with the terminal elimination half-life of vorapaxar.
-
In healthy volunteer studies, no changes in platelet P-selectin and soluble CD40 ligand (sCD40L) expression or coagulation test parameters (TT, PT, aPTT, ACT, ECT) occurred after single- or multiple- dose (28 days) administration of vorapaxar. No meaningful changes in P-selectin, sCD40L, or hs-CRP concentrations were observed in patients treated with vorapaxar in the phase 2/3 clinical trials.
-
Evaluation of Vorapaxar on QTc Interval: The effect of vorapaxar on the QTc interval was evaluated in a thorough QT study and in other studies. Vorapaxar had no effect on the QTc interval at single doses up to 48 times the recommended dose.
- Vorapaxar exposure increases in an approximately dose-proportional manner following single doses up to 16 times the recommended dose. Vorapaxar pharmacokinetics are similar in healthy subjects and patients.
- Absorption: After oral administration of a single vorapaxar 2.08 mg dose under fasted conditions, peak concentrations (Cmax) occur at 1 hour post-dose (range: 1 to 2 h). The mean absolute bioavailability as determined from a micro-dosing study is approximately 100%. Ingestion of vorapaxar with a high-fat meal resulted in no meaningful change in AUC with a small (21%) decrease in Cmax and delayed tine to peak concentration (45 minutes). Vorapaxar may be taken with or without food.
- Distribution: The mean volume of distribution of vorapaxar is approximately 424 liters (95% CI: 351-512). Vorapaxar and the major circulating active metabolite, M20, are extensively bound (≥99%) to human plasma proteins. Vorapaxar is highly bound to human serum albumin and does not preferentially distribute into red blood cells.
- Metabolism: Vorapaxar is eliminated by metabolism via CYP3A4 and CYP2J2. The major active circulating metabolite is M20 (monohydroxy metabolite) and the predominant metabolite identified in excreta is M19 (amine metabolite). The systemic exposure of M20 is ~20% of the exposure to vorapaxar.
- Elimination: The primary route of elimination is through the feces. In a 6-week study, 84% of the administered radiolabeled dose was recovered as total radioactivity with 58% collected in feces and 25% in urine. Vorapaxar is eliminated primarily in the form of metabolites, with no unchanged vorapaxar detected in urine.
- Vorapaxar exhibits multi-exponential disposition with an effective half-life of 3-4 days and an apparent terminal elimination half-life of 8 days. Steady state is achieved by 21 days following once-daily dosing with an accumulation of 5- to 6-fold. The apparent terminal elimination half-life for vorapaxar is approximately 8 days (range 5-13 days) and is similar for the active metabolite. The terminal elimination half-life is important to determine the time to offset the pharmacodynamic effect.
- Specific Populations: In general, effects on the exposure of vorapaxar based on age, race, gender, weight, and moderate renal insufficiency were modest (20-40%). No dose adjustments are necessary based upon these factors. Because of the inherent bleeding risks in patients with severe hepatic impairment, vorapaxar is not recommended in such patients.
- Drug Interactions
- Anticoagulants and antiplatelet agents: An interaction study with vorapaxar and warfarin in healthy subjects did not demonstrate a clinically significant pharmacokinetic or pharmacodynamic interaction. Vorapaxar did not affect prasugrel pharmacokinetics and prasugrel did not affect vorapaxar pharmacokinetics following multiple-dose administration at steady state. The pharmacokinetic interaction between vorapaxar and clopidogrel has not been evaluated. However, the use of vorapaxar on a background of clopidogrel is supported by the clinical data from TRA 2°P and TRA•CER.
- Effects of other drugs on vorapaxar: The effects of other drugs on the pharmacokinetics of vorapaxar were tested as change relative to vorapaxar administered alone (test-reference). Phase 3 data suggest that coadministration of a weak or moderate CYP3A inhibitor with vorapaxar does not increase bleeding risk or alter the efficacy of vorapaxar. No dose adjustment for vorapaxar is required in patients taking weak to moderate inhibitors of CYP3A.
- Effects of vorapaxar on other drugs: In vitro metabolism studies demonstrate that vorapaxar or M20 is unlikely to cause clinically significant inhibition or induction of major CYP isoforms or inhibition of OATP1B1, OATP1B3, BCRP, OAT1, OAT3, and OCT2 transporters. Specific in vivo effects on the pharmacokinetics of digoxin, warfarin, rosiglitazone and prasugrel were analyzed as a change relative to the interacting drug administered alone (test/reference). Vorapaxar is a weak inhibitor of the intestinal P-glycoprotein (P-gp) transporter. No dosage adjustment of digoxin or vorapaxar is required.
- Summarize the benefits and potential side effects of vorapaxar for the patient.
- Tell patients to take vorapaxar exactly as prescribed and not to discontinue vorapaxar without discussing it with the prescribing physician.
- Inform patients that they may bleed and bruise more easily and that they should report any unanticipated, prolonged or excessive bleeding, or blood in their stool or urine.
- Instruct patients to inform physicians and dentists that they are taking vorapaxar before any surgery or dental procedure and to tell the doctor performing any surgery or dental procedure to talk to the prescribing physician before stopping vorapaxar.
- Tell patients to list all prescription medications, over-the-counter medications, or dietary supplements they are taking or plan to take so that they physician knows about other treatments that may affect bleeding risk.
Indications
Dosing (Adult)
General Notes: Take with or without food
Black Box Warnings
Adverse Reactions
Overdose
Drug Interactions
Special Populations
Breasfeeding
Chemical Structure
Mechanism of Action
Pharmacodynamics
Pharmacokinetics
Counseling Points
MESH Terms & Keywords
|
|---|
|