Selegiline (Eldepryl, Zelapar): Drug Monograph
|
|---|
- Adjunct in the management of parkinsonian patients being treated with levodopa/carbidopa who exhibit deterioration in the quality of their response to this therapy
- General Notes:
- Eldepryl: Take in divided doses at breakfast and lunch.
- Zelapar: Take in the morning before breakfast and without liquid; avoid ingesting food or liquids for 5 minutes before and after taking the drug. Do not attempt to push through the foil backing; peel back the backing with dry hands, and gently remove the tablet (s). Immediately place on top of the tongue
- Parkinson's disease, Eldepryl:
- 10 mg/day by mouth as divided doses of 5 mg each taken at breakfast and lunch
- After 2-3 days of therapy, may attempt to reduce levodopa/carbidopa dose; during continued therapy, further reductions of levodopa/carbidopa may be possible
- Parkinson's disease, Zelapar:
- 1.25 mg by mouth once daily for at least 6 weeks
- After 6 weeks, may increase to 2.5 mg once daily if a desired benefit has not been achieved and the patient is tolerating therapy
- Max Dose: 2.5 mg by mouth once daily
- Maintenance: 1.25 mg or 2.5 mg by mouth once daily, depending on individual clinical response
- None (Eldepryl)
- Mild-moderate (Child-Pugh Score 5-9), Zelapar:
- Reduce daily dose (from 2.5 mg to 1.25 mg once daily), depending on clinical response
- Severe (Child-Pugh Score >9), Zelapar:
- Not recommended
- Known hypersensitivity to selegiline or any other components of the formulation
- Patients using meperidine, tramadol, methadone, or propoxyphene - serotonin syndrome can result
- Patients on any other MAO inhibitor (selective or non-selective) - increased risk for hypertensive crisis
- Patients using St. John's Wort, or cyclobenzaprine
- Patients using dextromethorphan - may result in episodes or=f psychosis or bizarre behavior
- Hypertension
- Falling asleep during activities of daily living and somnolence
- Dyskinesia
- Hallucinations/psychotic-like behavior
- Impulse control/compulsive behaviors
- Withdrawal emergent hyperpyrexia and confusion
- Melanoma
- Irritation of the buccal mucosa
- Do not exceed recommended dose of 10 mg/day - risks associated with non-selective inhibition of MAO (i.e., "cheese reactions")
- Exacerbation of levodopa—associated side effects with patients on adjunctive therapy - reduce dose of levodopa by approximately 10-30%
- Severe live or kidney dysfunction - use with caution. No information available on selegiline or its metabolites in these patients
- Patients with peptic or duodenal ulcer, labile hypertension, cardiac arrhythmias, severe angina pectoris, or psychosis - use with caution as treatment may exacerbate these conditions
- Transient increases in liver transaminases may occur with treatment
- Impulse control disorders and compulsions - pathological gambling, increased libido, hypersexuality, binge eating, shopping and different kinds of compulsive/repetitive activities may occur
- Abnormal movements such as dyskinesias
- Nausea
- Agitation
- Confusion
- Hallucinations
- Impulse control/compulsive behaviors
- Headache
- Postural hypotension
- Cardiac arrhythmias
- Vertigo
- Micturition difficulties
- Skin reactions
- Irritation of the buccal mucosa
- No overdosage cases known.
- Symptoms may include disorders of the CNS and cardiovascular systems, such as drowsiness, dizziness, faintness, irritability, hyperactivity, agitation, severe headache, hallucination hypertension, hypotension, vascular collapse, rapid and irregular pulse, precordial pain, respiratory depression and failure, hyperpyrexia, diaphoresis
- Treatment is symptomatic and supportive
- Pethidine - serious, sometimes fatal reactions with concomitant use or in patients who have discontinued MAOl therapy and are then started on Pethidine within two weeks of discontinuation. Agitation, delirium irritability, hyperpyrexia, restlessness, rigidity, stupor, and sweating may result.
- Selective serotonin reuptake inhibitors (SSRIs) - Serious, sometimes fatal, reactions with concomitant use and in patients who have recently discontinued SSRIs (particularly fluoxetine) and then started on a MAOl. Agitation, ataxia, cold sweats, hypertension, mania, pseudophaecochromocytoma, "serotonergic reaction" and shivers may result. Allow at least five weeks between discontinuation a SSRI and initiation of selegiline therapy; two week period between selegiline discontinuation and SSRI initiation
- Monoamine oxidase inhibitors - no tolerability problems, but the exogenous amine sensitivity factor may increase. Use a diet low in exogenous amines such as tyramine
- Non-selective MAOls (linezolid) - may cause severe hypertension or hypotension
- Serotonergic drugs - uncommon symptoms may include myoclonus, tremor, confusion, restlessness, ataxia and hyperreflexia. Usually short-lived, but can lead to intensive care admissions and may be fatal. Discontinue drugs responsible
- Tricyclic antidepressants - severe CNS toxicity may result, including tremor, agitation and restlessness, unresponsiveness, hyper/hypotension, dizziness, diaphoresis, seizures and changes in behavioral and mental status.
- Oral contraceptives (tablets containing the combination of gestodene/ethinyl estradiol or levonorgestrel/ethinyl estradiol)- may increase the oral bioavailability of selegiline. Use caution
- Pregnancy: Pregnancy Category C
- Labor and Delivery: None
- Nursing Mothers: It is not known whether the drug is excreted in human milk. Avoid use
- Renal Impairment: Orally disintegrating tablet not recommended for severe impairment
- Hepatic Impairment: Orally disintegrating tablet dose adjustment for mild to moderate impairment; not recommended for severe impairment
- Pediatric Patients: The safety and effectiveness have not been evaluated.
- Geriatric Patients: The overall incidence of adverse reactions may increase in geriatric patients compared to non-geriatric patients. Risk for hypertension and orthostatic/postural hypotension may be greater
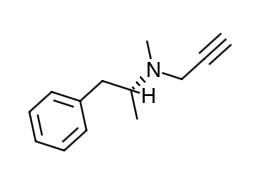
- Scientific Name: (-)-(R)-N, α-dimethyl-N-2-propynylphenethylamine hydrochloride
- Empirical Formula: C13H17N•HCl
- Molecular Weight: 223.75
- Selegiline is an irreversible inhibitor of monoamine oxidase (MAO), which regulates the metabolic degradation of catecholamines and serotonin in the CNS and peripheral tissues. At recommended doses, selegiline is selective for MAO type B (MAO-B), the major form in the brain. Inhibition of MAO-B activity, by blocking the catabolism of dopamine, may result in increased dopamine levels; however, there is evidence that selegiline may act through other mechanisms to increase dopaminergic activity.
- A pharmacodynamic study investigating daily doses of 2.5 mg, 5 mg, and 10 mg for tyramine sensitivity showed that increased tyramine sensitivity resulting in increased blood pressure (because of MAO-A inhibition and decreased selectivity for MAO-B) occurred with dosing above the recommended level (2.5 mg daily). An increase in tyramine sensitivity for blood pressure responses appears to begin at a dose of 5 mg selegiline daily.
- Absorption: Selegiline is readily absorbed from the gastrointestinal tract. The maximal concentrations are reached in 0.5 hours after oral administration. The bioavailability is low-on average 9.4 ± 5.9% of unchanged selegiline from a 10 mg oral dose reaches the systemic circulation. A substantial increase in selegiline bioavailability (up to threefold) occurs when selegiline is administered with food high in fat. Zelapar: Selegiline disintegrates within seconds after placement on the tongue and is rapidly absorbed. Detectable levels of selegiline have been measured at 5 minutes after administration, the earliest time point examined. Selegiline is more rapidly absorbed from the 1.25 or 2.5 mg dose (Tmax range: 10-15 minutes) than from the swallowed 5 mg selegiline tablet (Tmax range: 40-90 minutes). Mean (SD) maximum plasma concentrations of 3.34 (1.68) and 4.47 (2.56) ng/mL are reached after single dose of 1.25 and 2.5 mg disintegrating tablet, compared to 1.12 ng/mL (1.48) for the swallowed 5 mg selegiline tablets (given as 5 mg bid). On a dose-normalized basis, the relative bioavailability of selegiline from disintegrating tablet is greater than from the swallowed formulation. The pre-gastric absorption from the disintegrating tablet and the avoidance of first-pass metabolism results in higher concentrations of selegiline and lower concentrations of the metabolites compared to the 5 mg swallowed selegiline tablet. Plasma Cmax and AUC of the disintegrating tablet were dose proportional at doses between 2.5 and 10 mg daily.
- Food effects: When the disintegrating tablet is taken with food, the Cmax and AUC of selegiline are about 60% of those seen when taken in the fasted state. Since the disintegrating tablet is placed on the tongue and absorbed through the oral mucosa, the intake of food and liquid should be avoided 5 minutes before and after administration.
- Distribution: Up to 85% of plasma selegiline is reversibly bound to proteins.
- Metabolism: Following a single dose, the median elimination half-life of selegiline was 1.3 hours at the 1.25 mg dose. Under steady-state conditions, the median elimination half-life increases to 10 hours. Upon repeat dosing, accumulation in the plasma concentration of selegiline is observed both with the disintegrating tablet and the swallowed 5 mg tablet. Steady state is achieved after 8 days. Selegiline is metabolized in vivo to l-methamphetamine and N-desmethylselegiline and subsequently to l-amphetamine; which in turn are further metabolized to their hydroxymetabolites. The disintegrating tablet also produces a smaller fraction of the administered dose recoverable as the metabolites than the conventional, swallowed formulation of selegiline. In vitro metabolism studies indicate that CYP2B6 and CYP3A4 are involved in the metabolism of selegiline. CYP2A6 may play a minor role in the metabolism.
- Elimination: Following metabolism in the liver, selegiline is excreted primarily in the urine as metabolites (mainly as l-methamphetamine) and as a small amount in the feces.
- Special Populations
- Age: The effect of age on the pharmacokinetics of selegiline following administration has not been adequately characterized.
- Gender: There are no differences between male and female subjects in overall (AUC∞) time to maximum exposure (Tmax) and elimination half-life (t1/2) after administration. Female subjects have an approximate 25% decrease in Cmax compared to male subjects. However, since the overall exposure (AUC∞) is not different between the genders, this pharmacokinetic difference is not likely to be clinically relevant.
- Race: No studies have been conducted to evaluate the effects of race on the pharmacokinetics of the drug.
- Renal Impairment: Following once-daily dosing of 2.5 mg to selegiline steady-state (10 days) in 6 subjects with mild renal impairment (ClCr >50 to 89 mL/ min) and in 6 subjects with moderate renal impairment (ClCr >30 to 50 mL/min),AUC and Cmax of selegiline and desmethylselegiline were not substantially different from healthy subjects; however, methamphetamine and amphetamine exposures were increased by 34-67% in subjects with moderate renal impairment. Following once-daily dosing of 1.25 mg to steady-state (10 days) in 6 end-stage renal disease patients, off dialysis, selegiline exposure was not substantially different from that in healthy subjects, however methamphetamine and amphetamine exposures were increased approximately 4-fold compared to healthy subjects.
- Hepatic Impairment: Subjects with mild hepatic impairment (Child-Pugh score 5 to 6), received once-daily dosing of 2.5 mg to selegiline until they attained steady-state (10 days). The AUC and Cmax of selegiline were 1.5-fold higher and the AUC and Cmax of the metabolite desmethylselegiline were 1.4-fold and 1.2-fold higher. In subjects with moderate hepatic impairment (Child-Pugh score 7 to 9), the AUC of selegiline and desmethylselegeline increased 1.5-fold and 1.8-fold, respectively, whereas the Cmax of selegiline and desmethylselegiline were comparable to healthy subjects. Patients with severe hepatic impairment (Child-Pugh score >9) had a 4-fold increased AUC of selegiline, 3-fold increased Cmax of selegiline, a 1.25-fold increased AUC of desmethylselegeline and 50% reduced Cmax of desmethylselegeline. Methamphetamine and amphetamine metabolite AUC values were not affected by liver dysfunction.
- Drug Interactions:
- No studies have been conducted to evaluate drug interactions on the pharmacokinetics of selegiline. Effect of CYP3A inhibitor itraconazole: Itraconazole (200 mg QD) did not affect the pharmacokinetics of selegiline (single 10 mg oral, swallowed dose). Although adequate studies to investigate the effect of CYP3A4-inducers on selegiline have not been performed, drugs that induce CYP3A4 (e.g., phenytoin, carbamazepine, nafcillin, phenobarbital, and rifampin) should be used with caution.
- Drug Interaction Studies: No drug interaction studies have been conducted to evaluate the effects of other drugs on the pharmacokinetics of selegiline or the effect of selegiline on other drugs. In vitro studies have demonstrated that selegiline is not an inhibitor of CYP450 enzymes. Selegiline and two of its metabolites, methamphetamine and desmethylselegiline, have little or no potential to induce CYP1A2 and CYP3A4/5 under clinical conditions.
- Advise patients of the possible need to reduce levodopa dosage after initiation of therapy
- Instruct patients not to exceed 10 mg/day and explain the risk of using higher daily doses; provide a brief description of the "cheese reaction"
- Inform patients about the signs and symptoms associated with MAOl-induced hypertensive reactions; instruct patients to immediately report to their physician any severe headache or other atypical/unusual symptoms not previously experienced
- Instruct patients to inform their physician if new or increased gambling urges, increases sexual urges, or other intense urges develop
- Advise patients to avoid certain food (e.g., aged cheese) containing a very large amount of tyramine while on therapy
- Instruct patients to inform their physician if taking or planning to take any prescription or OTC drugs
- Instruct patients not to drive a car or engage in other potentially dangerous activities until the patient has gained sufficient experience with therapy
- Inform the patient that symptomatic (or asymptomatic) hypotension may develop; caution against rising rapidly after sitting or lying down for prolonged periods and at the initiation of treatment
- Advise patients to have periodic skin examinations
- Inform patients that the drug may cause irritation of the buccal mucosa, and that it contains aspartame, which could cause problems in patients with phenylketonuria
- Advise patients to contact their healthcare provider if they wish to discontinue treatment or decrease the dose of the drug
Indications
Dosing (Adult)
Hepatic Dosing
Contraindications
Warnings
Adverse Reactions
Overdose
Drug Interactions
Special Populations
Chemical Structure
Mechanism of Action
Pharmacodynamics
Pharmacokinetics
Counseling Points
MESH Terms & Keywords
|
|---|
|